Hello everyone!
My name is Grady Day, and I'm a senior at BASIS Scottsdale. In this blog, I'll be documenting my Senior Research Project over the next few months. At BASIS Scottsdale, seniors spend their last trimester interning and performing research, ending with a presentation in May. Here's some more information about and examples of Senior Research Projects at BASIS Scottsdale. Specifically, my project will be focusing on the genetics of multiple myeloma, and I'll be working at the Mayo Clinic in Scottsdale.
But before I start talking about my project, I’ll introduce myself and my interests briefly.
After begging for a microscope for my sixth birthday, seeing my own red blood cells ebb and flow across the slide helped spark a lifelong interest in using biology to explain the world around me. Ever since then, I’ve loved every aspect of biology. Activities like Science Bowl have helped me focus my interests in biology, reinforcing my desire to understand the intricate molecular interactions that make up life. In fact, I’m taking a short break from studying for Science Bowl to write this post—my final Regional-level competition is tomorrow, and it’ll be my first as team captain.
This summer, I had the opportunity to be a part of research on bacterial competition at UC Santa Barbara. It was my first real lab experience, and I loved every part of it. I got to actually perform the techniques I’d learned about in class, and I was amazed when I saw the results of my first PCR and gel electrophoresis. Being able to actually visualize the fundamental molecular processes that make up life was incredibly exciting, and it’s made me look forward to studying biology in college.
Now, I’m excited to build upon and expand my experience by working in a cancer genetics lab. I'll be starting with cell lines to develop a method for identifying a certain translocation, and then moving towards applying that method to real patient samples in order to characterize the effect of that transolcation on patient oucomes. I’m only barely getting started on my project, so I’ll start posting in more detail about the specifics very soon. Each week, I’ll be blogging about what I’ve done in the lab and what progress I’ve made. If you’re interested in reading about some other projects from some of my classmates, check out their blogs through the links on the right. I hope you’re as excited about this project as I am! I’ll post again next week!
Thanks for reading,
Grady Day
Week 10
Hi everyone!
I can’t believe it’s been 10 weeks already! Luckily, it
looks like I’m just about finished putting together the results for my project.
On Monday, I received my first set of patient samples. These
samples came from a single patient at 3 distinct timepoints. Although they were
collected as bone marrow samples, I only worked with the extracted DNA leftover
from when our lab had sent these samples out for sequencing. To understand what
information we can gain from these samples, we first need to know what makes
these samples different.
The first sample was collected very early in the disease’s
progression, called smoldering multiple myeloma. I was surprised to learn that
the distinction between SMM, the preceding stage (called monoclonal gammopathy
of undetermined significance, or MGUS), and full blown multiple myeloma is
actually based solely on a few ratios. The difference between a MGUS, a pre-clinical
condition that requires no treatment and only semi-annual blood tests for
monitoring, and SMM, a condition usually treated aggressively via a combination
of a bone marrow transplant and several drugs, is based only on whether a bone
marrow sample contains more or less than 10% plasma cells.
The distinction between multiple myeloma and MGUS/SMM,
however, is much clearer. By time the disease has progressed to MM, the malignant
plasma cells have displaced far more cells in the bone marrow. Therefore, while
the first sample I’m working with consists largely of healthy cells normally
found in bone marrow along with some tumor cells, the second sample I have is predominantly
myeloma cells.
The final sample I have was collected after treatment with a
combination therapy of two drugs. Because these therapies have killed off many
of the myeloma cells in the bone marrow, the final bone marrow sample may have
even less myeloma cells than the original SMM sample. That doesn’t mean the therapy
will be effective long-term, though, because these surviving cells may be
different from the original population of tumor cells in the SMM bone marrow. Recent
studies show this actually occurs through natural selection—one cell in the
initial SMM population gets a mutation that lets it reproduce more rapidly,
leading to it overtaking the growth of other tumor cells in the initial population.
Our goal is essentially to determine what mutations are
found in the full-blown MM sample but not the other two—and as soon as I get
the sequencing data back for these samples, I’ll be able to design a qPCR assay
that will help us in better detecting these differences.
Even though the progress I’ve made is far from
revolutionary, I’m incredibly glad to see how much I’ve learned since my first
day in the lab. I’m excited to see the other ways I’ll be able to apply this assay
(and the other projects I will be involved with) throughout the summer! Thank
you all for following my project!
Hi everyone!
This week I’ve really started getting some great results to report!
On Monday, I was able to order the first qPCR assay for L363, the cell line I
had sequenced last week.
The qPCR setup I’m using is a lot like normal PCR in that I
ordered two primers (one forward and one reverse) that span about 150 base
pairs across the VJ breakpoint. I’m even using basically the same Taq
polymerase in my reactions, which luckily makes it very easy for me to optimize
my qPCR reactions first using regular PCR + gel electrophoresis. The only new ingredient
I added to the qPCR is an intercalating dye called SYBR green. Intercalating basically
means that it binds to double-stranded DNA, and when it does bind DNA it fluoresces
green when exposed to blue light. The intensity of the green light from the
SYBR green is actually directly proportional to the quantity of DNA currently
in each tube. With PCR, we start with a very low quantity of the template DNA,
containing anywhere from a few hundred thousand to a few copies of the IgL
breakpoint we’re interested in. Each subsequent cycle the qPCR goes through
essentially doubles the number of the target sequence without amplifying the
rest of the template. The overall effect of this is that when you plot the
fluorescence levels vs cycle number, you basically find out that every subsequent
fluorescence level is roughly 2x the intensity in the last cycle.
Using a few samples of known dilutions, we can prepare what’s
called a standard curve for this particular qPCR assay, essentially telling us the
relationship between fluorescence levels and initial template DNA
concentration. I’m probably making this sound a lot more difficult than it really
is, because all the calculations are basically done automatically by the
software we use. We can work backwards
from this to figure out how many copies of the IgL VJ breakpoint of L363 were
present in each sample reaction we are interested in running.
Designing and testing these qPCR assays is definitely my favorite
aspect of the research process so far. is probably my favorite part of my project so
far. However, I’m going to have to figure out the best conditions pretty
quickly before I move onto real patient samples. Now, because I’m working with
a cell line, I had nearly complete control over how much DNA I had put into
each reaction, and a basically unlimited supply of DNA to work with. With
patient samples, though, I’ll have only a few tries to get it right before I use
them all up—so right now I’m going to focus pretty heavily on optimizing as
fast as I can.
Thanks for reading!
Lessons Learned
Hi everyone!
I’ve spent lots of time in the last week playing around with the new downstream primers I ordered at the end of last week. I’m not 100% satisfied yet, but I’m very confident I’ll be there soon enough. I am starting to feel the time pressure of finishing in time for the presentation, and I think because of that some of my work has started to get slightly rushed if not sloppy.
In a lot of ways, what I’ve been doing is basically a repeat of what I already did with the first set of primers I’ve ordered. I’ve tried lots of different primer combinations and various reaction conditions, and it’s definitely helped significantly so far. The new primer set worked on one of the cell lines I’m working with right away, so I sent that product off for sequencing right away. An now, after just under a week of optimization, I think I’m getting a decently clean product on all 5 cell lines. I got an excellent read (~450 base pairs in length, which is near the upper limit for Sanger sequencing) on the sequencing for the product I sent out. That’s excellent news, because I now have more than enough of the rearrangement sequenced than what I need to design a qPCR assay. If I absolutely had to, I could do it with only ~75 base pairs on each side of the breakpoint, but it’ll be significantly easier with the ~200 base pairs on each side I have to work with now.
But because I’ve been trying to move forward as quickly as possible, I;ve started noticing I’m making some unnecessary mistakes. For example, I accidentally forgot to tighten the caps on all the reaction tubes before placing them in the thermocycler. That’s a big problem because PCR cycles alternate between ~60 to ~95ºC, meaning the water inside the PCR tube can evaporate if the caps aren’t on tight enough. When I took them out the next morning after the PCR cycles were finished, most of the reactions were completely ruined, now only a hard, green resin that’s totally useless. And because that was the nested reaction, it means I needed to repeat both the external and nested reactions in order to try that specific experiment again.
Luckily, thanks to the cheaper Taq polymerase I’ve been using, little mix-ups like these aren’t costing the lab in any significant way. I have learned an important lesson, however: for lab work, it’s always better to go slowly and carefully. Even if I get lucky and don’t make a mistake, having sloppy/incomplete notes for an experiment means that at best I’ll need to spend more time interpreting the results and at worst I’ll need to repeat it completely.
Going forward, I won’t stop trying to move forward with my project as quickly as possible, but I will be sure to do everything much more carefully.
Thanks for reading!
Week 7
Hi everyone!
I’ve had a very exciting week! While waiting for the
sequencing results to come in, I got an introduction to several ongoing
projects related to my own, focused on different approaches towards identifying
the processes that are misregulated in myeloma cells.
The project I’ve had the most time to work on so far makes
use of an analysis technique called mate pair sequencing, a powerful technique
for identifying changes in the structure of chromosomes. Some of these
structural variations (SVs) are already known to be major driving factors of
oncogenesis, especially in multiple myeloma. SVs that bring oncogenes near enhancers
or super-enhancers (DNA sequences that turn on nearby genes) are thought to
have a major influence on how myeloma can progress at very different rates in
patients. SVs involving a particular oncogene called Myc are a topic the
Bergsagel lab has studied extensively using mate pair sequencing and other
techniques. Currently, Myc translocations have been identified in the vast
majority of myeloma patient samples and cell lines. Previous research shows
that Myc translocations occur very late in tumor progression. Our working hypothesis
is that Myc translocations might be what separates patients whose cancers never
progress beyond a pre-cancerous stage called MGUS from patients who progress
rapidly into full-blown MM.
To test this hypothesis, we need to search many patient
samples taken at several timepoints for Myc translocations. Mate pair is excellent
for screening the entire genome for SVs, but it’s expensive, slow, and requires
relatively large quantities of very well-preserved patient samples. Analyzing mate
pair data requires making some judgement calls on whether some ambiguous reads
represent real SVs, so it can miss certain SVs. Instead, by analyzing Myc
expression directly, we can more precisely identify samples with Myc translocations.
Because IgL rearrangements are generated randomly in pre-B cells, each IgL
rearrangement will be unique to a particular lineage of plasma cells (called a
clone). In any single patient, the myeloma/MGUS lesions are not monoclonal, meaning
a single tumor sample actually consists of a mix of different clones. The qPCR
assays I’m designing will let us quantify the proportion of a sample consisting
of a particular clone, letting us compare Myc expression levels to the relative
abundance of the clone in question.
On Thursday, the sequencing results came in. Although the sequencing
data was excellent in terms of clarity, the primer set I had used didn’t cover enough
of the J end of the VJ rearrangements, meaning we can’t generate the qPCR
assays just yet. To fix this, I ordered new J primers that anneal further
downstream, meaning the amplicon should be long enough to determine the entire
full sequence of the VJ rearrangement. Hopefully, by next Thursday we’ll have
the new sequencing results, and from these I’ll be able to design an initial qPCR
assay!
Thanks for reading!
Week 6
Hi everyone! I can’t believe six weeks have already gone by! I’m happy to report that I’ve made some really substantial progress in the past week.
In last week’s post, I wrote that I’d be using primers from an old paper about IgL rearrangements in a technique called nested PCR.
 |
| http://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-methods.html |
Nested PCR is really useful for reducing the impact of non-specific binding of primers to template DNA. Primer sequences don’t actually have to match the target sequence perfectly and even with several mismatches they can often still anneal to the template strand. And throughout the genome, it’s often the case that multiple forward and reverse primer binding sites are located sufficiently close to each other to allow for amplification of several off-target sequences.
Here’s where the nested part comes in. We used a set of external, or outer, primers that were really similar to the ones I had designed, and they gave us pretty much the same kind of messy, largely nonspecific product as my earlier primers had. In nested PCR, that often-messy product from the first reaction is used as the template DNA for the second reaction. The forward primer for the nested reaction anneals slightly downstream from the forward external primer, and the reverse primer from the nested reaction anneals slightly upstream from the external reverse primer.
Because the nested PCR reaction uses already-amplified DNA as the template, it drastically cuts down on the number of potential binding sites for forward and reverse primers. This means that nested PCR allows for much less off-target amplification than traditional PCR. Here’s the external products run out on a gel.

You can see it’s really not much cleaner than what I had before. However, after the nested reaction, the difference is very clear.
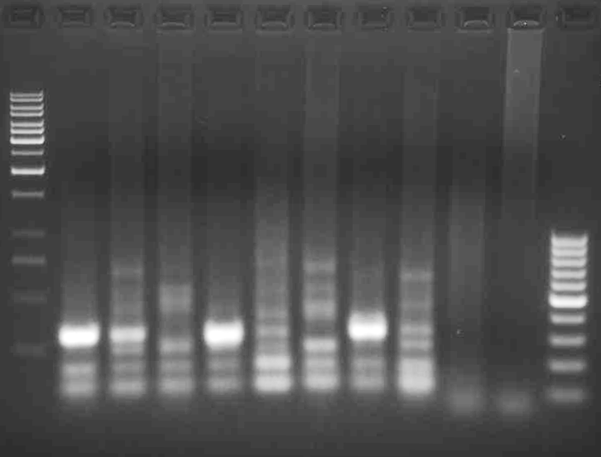
This is 10 separate reactions using the XG2 external PCR product along with different nested primers in order to determine which primer pair will work best for sequencing. You can see there is a single clearly visible dominant band in three of the lanes (1, 4, and 7). That quantity of product is more than sufficient for sequencing, which is the next step.
This nested PCR technique isn’t perfect, because it takes about 7 hours to run completely, and requires many reactions. That being said, I’m not convinced there’s a better alternative, and these disadvantages really don’t amount to much more than minor inconveniences. I am still working on optimizing this reaction for annealing temperature and other reaction conditions
In addition, I’ve tried using a different enzyme. For all my previous reactions, I had been using a high-fidelity polymerase called Q5, which costs roughly $2 per reaction. This enzyme, like many other high-fidelity polymerases, is really useful for amplifying very long products with very few errors, so it’s the first enzyme my lab turns to for PCR. For this application, I really don’t need anything that fancy. Instead, I’m using a more basic Taq polymerase that only costs ~$0.10 per reaction. This week, with all the nested reactions I was running, I needed as many as 60 PCRs per day, so reduced reagent costs are pretty substantial. The Taq master mix is also a nice time-saver, because it comes pre-mixed with a loading dye that allows for very easy gel electrophoresis.
Interestingly, although I expected this to only be a cost-saving measure, it drastically improved the specificity of my PCRs. Dr. Riggs and I think this is probably due to the Q5 enzyme + buffer being more tolerant of primer/template mismatches. Here’s the same nested PCR reactions, but using the Taq enzyme instead of the high-fidelity Q5.
 |
| SECURE THE BAND ALERT: it doesn't get much clearer than these! |
I’m planning on sending these out for sequencing on Monday, and hopefully I’ll be able to design and try my first qPCR by late next week!
Thanks for reading!
Spring Break!
Hi everyone! I hope you’ve all enjoyed your spring breaks so
far--I know I have! Last week I wrote about some of the different reactions I’ve
tried with the primers I’d ordered. This week I had time to pretty much finish most
of those tests. Unfortunately, I haven’t been able to get the results I’d hoped
to see.
My goal was to get one single amplicon from each PCR, and
you might remember from the picture I shared last week that at least four or
five were consistently appearing. I’ve
tried a few different approaches, including varying the reaction temperature, primer
concentrations, and template genomic DNA concentration. I saw some slight
improvements, but never anything close to a single band on the gels I ran.
Now, having an initial PCR product with multiple bands isn’t
an insurmountable barrier to moving forward with qPCR assay design. But having
many bands in that first PCR product means it’ll be hard to tell if the correct
band is present. And if the correct band isn’t present, that means when we send
the PCR product in for sequencing we won’t know if we’ll get the correct
result. So, while we could live with not getting a single clean result from the
initial PCR, we have to be sure the correct product is in there somewhere.
I used two methods to determine whether the correct amplicon
was present. The first was a restriction enzyme digest. Restriction enzymes are
proteins that recognize a short sequence of DNA and cleave the DNA strand at that
point. They are very useful for creating recombinant DNA by splicing together genes
from different sources. In my case, though, I just needed to make a cut at a
known location in the amplicon I predicted. I found a cleavage site of an enzyme
called BglII that would cut the correct amplicon into two pieces of different
lengths. I set up a digest on my PCR product with BglII, and then ran the digest
next to the original product. If my original product was correct, then I should
have seen the longer band disappear and be replaced by two shorter bands. When
I actually ran the digest, though, I didn’t get a clear result.
The other method I tried is called nested PCR. Nested PCR
uses the product from one PCR as the template for another PCR, this time using
primers that anneal within the original product. I used two primers that would
anneal inside the correct product to set up a few PCRs. The results from those
were also inconclusive, unfortunately.
I haven’t come close to exhausting all the possibilities for
testing my primers. I could spend months trying to get these primers to work.
However, I can probably be more productive by trying a different approach. I
told you before that I hadn’t found any prior research containing primers I
could use, but that’s not entirely true. Originally, I was hoping to find
primers that would let determine which variable and joining regions were rearranged
using only a single reaction. However, there is a paper with a set of nested
primers (meaning two reactions will be required) for amplifying IgL breakpoints.
Dr. Riggs helped me order that paper’s primers, and they should arrive next
Tuesday. That paper showed some impressive success rates for amplifying IgL
rearrangements, and all the simulations I’ve ran confirm those primers should
amplify all rearrangements. I’m excited to try those out! Once I can amplify these
initial rearrangements, I can move towards qPCR assay design, which will let me
quantify these rearrangements in patient samples.
Enjoy the rest of your break!
Subscribe to:
Comments (Atom)